
From Boom to Regulatory Bust? The Evolving Landscape for Rare Genetic Disease Therapies
Explore the volatile world of rare disease biotechs in 2025, from Novartis's $12B Avidity acquisition sparking optimism to FDA scrutiny causing stock plunges for uniQure and Sarepta.
In the fast-evolving landscape of biotechnology, the rare disease sector—particularly those focused on genetic and neuromuscular disorders—has long been a beacon of hope for patients with limited treatment options. Advances in gene editing, RNA therapeutics, and platform technologies have promised transformative therapies for conditions like Duchenne muscular dystrophy (DMD), Huntington's disease, and spinocerebellar ataxia (SCA). Yet, recent events suggest a turbulent phase, where regulatory scrutiny, trial setbacks, and market volatility are testing the resilience of companies and investors alike. This article delves into the latest developments, drawing from press releases, FDA communications, and sector reactions, to provide a comprehensive view of the challenges and potential paths forward.
The Spark: Novartis's Bold Acquisition and Initial Market Surge
The momentum appeared to build on October 26, 2025, when Novartis announced its agreement to acquire Avidity Biosciences (NASDAQ: RNA) for approximately $12 billion, or $72 per share—a 46% premium over the prior closing price. This deal, expected to close in early 2026 pending approvals, underscores Novartis's commitment to strengthening its neuroscience pipeline with Avidity's innovative RNA therapeutics platform. Avidity's lead candidate, AOC 1001 (now delpacibart etedesiran), targets myotonic dystrophy type 1 (DM1) and has shown promising Phase 1/2 results, including reductions in myotonia and improvements in muscle function. The acquisition also bolsters Novartis's portfolio in neuromuscular diseases, complementing its existing spinal muscular atrophy (SMA) gene therapy, Zolgensma.
This news ignited short-lived optimism across the sector. Stocks of similar clinical-stage biotechs surged: Dyne Therapeutics (NASDAQ: DYN) jumped over 20%, PepGen (NASDAQ: PEPG) saw gains, Wave Life Sciences (NASDAQ: WVE) followed suit, and even Solid Biosciences (NASDAQ: SLDB)—a company with a focus on DMD gene therapy—experienced a brief uplift. Sarepta Therapeutics (NASDAQ: SRPT), despite its ongoing controversies, initially benefited from the ripple effect. Analysts viewed this as a potential rally for gene-editing and oligonucleotide-based platforms, signaling investor appetite for innovative therapies in underserved rare disease spaces.
However, Sarepta's year had already been marred by safety concerns surrounding its DMD gene therapy, Elevidys, which faced scrutiny over patient deaths and efficacy questions despite being the first commercialized product in this class. This set the stage for a dramatic reversal.
The Headwinds: Regulatory Shifts and Trial Setbacks
Just as enthusiasm peaked, a cascade of negative announcements reversed the tide, coinciding with broader market declines. On November 3, 2025, uniQure (NASDAQ: QURE) issued a press release revealing unexpected FDA feedback during a pre-Biologics License Application (BLA) meeting for AMT-130, its investigational gene therapy for Huntington's disease. The FDA indicated that the current Phase 1/2 dataset is insufficient for BLA submission, requiring a Phase 3 confirmatory trial—a stark departure from guidance provided in November 2024, which had supported an accelerated approval pathway. uniQure's CEO, Matt Kapusta, expressed surprise: "We are surprised by the FDA's feedback... which is a drastic change from the guidance the FDA provided in November 2024." Key figures from the PR include 39-month data showing a dose-dependent slowing of disease progression (measured by composite Unified Huntington's Disease Rating Scale, or cUHDRS) and reductions in neurofilament light chain (NfL) levels, a biomarker of neurodegeneration. Despite these positives, the stock plummeted over 50%, erasing recent gains and heightening sector-wide anxiety.
The following day, November 4, 2025, Sarepta released its Q3 earnings, compounding the gloom. While sales exceeded expectations at $386 million (driven by $240 million from Elevidys), the company disclosed that its exon-skipping therapies, Vyondys 53 and Amondys 45, failed to meet the primary endpoint in the confirmatory EMBARK trial. The North Star Ambulatory Assessment (NSAA) score showed no statistically significant improvement over placebo. This failure raises questions about full approval conversions from accelerated pathways and echoes ongoing safety issues, including prior patient deaths linked to Elevidys. The stock dove over 35%, reflecting investor disillusionment.
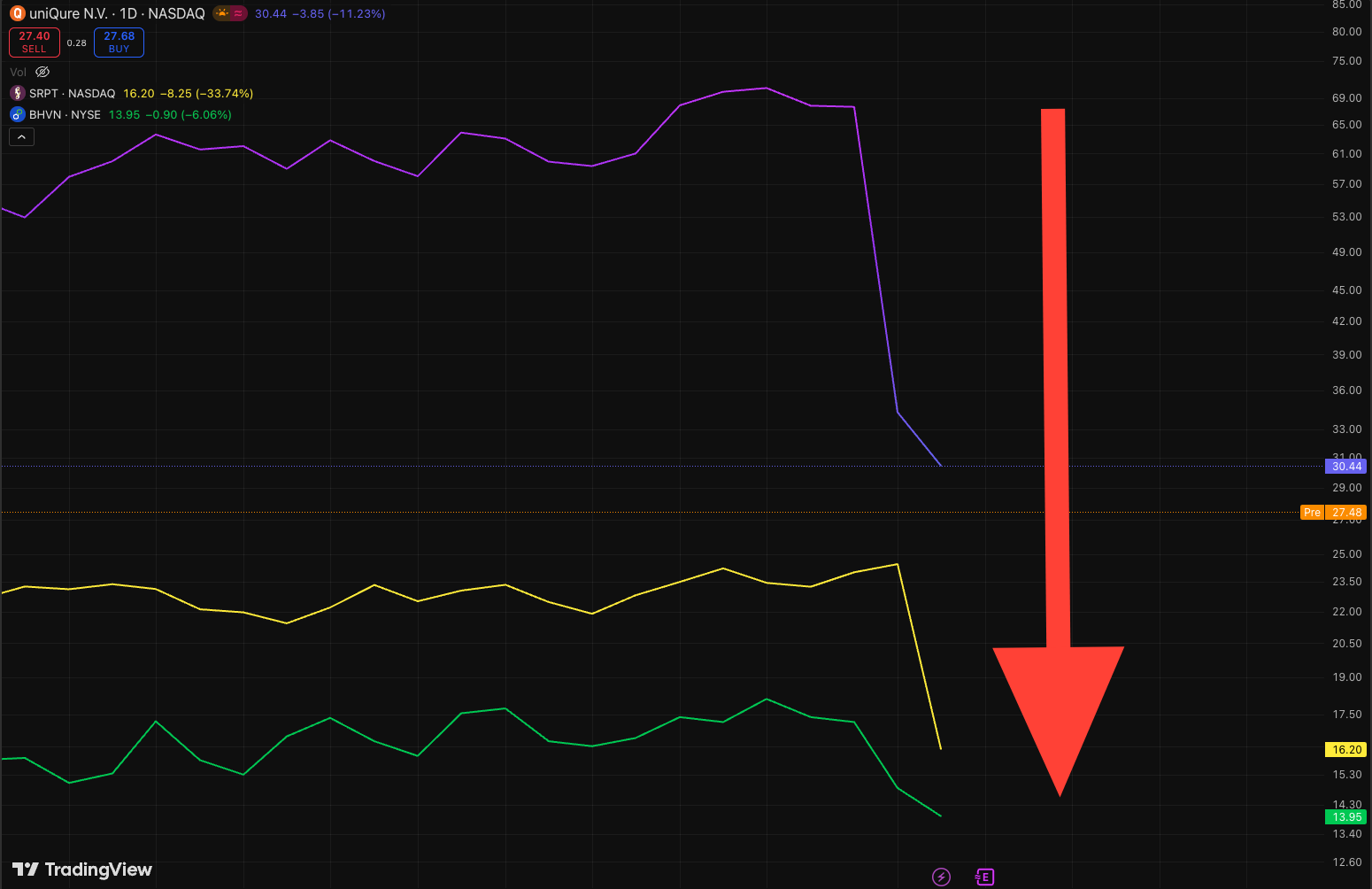
Simultaneously, Solid Biosciences reported its Q3 results on November 3, 2025, with mixed reception. Interim data from the INSPIRE DUCHENNE trial for SGT-003—a next-generation DMD gene therapy—were encouraging: As of September 29, 2025, 23 participants dosed showed functional improvements, including a mean 6.5-point increase in NSAA scores at 6 months and robust microdystrophin expression (up to 90% of muscle fibers). The therapy was generally well-tolerated with a steroid-only regimen, addressing prior cardiac safety concerns. However, the company announced plans to meet with the FDA in H1 2026 to discuss pathways, including accelerated approval—implying a delay from earlier timelines. Amid the sector's downturn, market excitement was muted, with the stock dipping despite the positives.
Adding to the week's turmoil, Biohaven (NYSE: BHVN)—though less directly tied to gene editing—received a CRL from the FDA on November 5, 2025, for VYGLXIA (troriluzole) in SCA. The agency cited inadequate evidence from the trial design, following a prior PDUFA delay. Troriluzole had shown 50-70% slowing of disease progression in Phase 3 data, but the CRL highlights persistent issues with pivotal trial alignments—previously assumed to be FDA-sanctioned.
FDA's Stance: Balancing Innovation and Caution
These events reflect the FDA's evolving approach to rare disease therapies. A recent Biospace article highlights the agency's efforts to accelerate gene-editing reviews, with CBER Director Peter Marks planning a paper on flexible mechanisms for "bespoke therapies" in ultra-rare conditions. This includes platform approvals for gene editors, inspired by CRISPR successes like the treatment of CPS1 deficiency. However, the FDA has revoked designations (e.g., Sarepta's Platform Technology Designation in July 2025 due to deaths) and emphasized confirmatory trials.
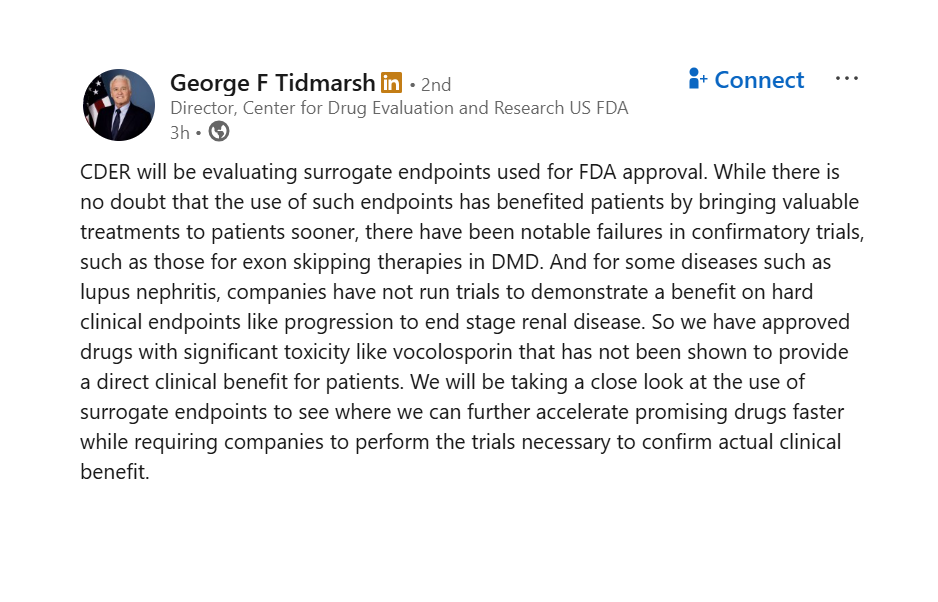
Dr. George Tidmarsh's September 2025 LinkedIn post (later deleted) encapsulates this scrutiny: "We have approved drugs with significant toxicity like voclosporin [Lupkynis] that has not been shown to provide a direct clinical benefit for patients." He also noted "notable failures of surrogate endpoint-based confirmatory trials such as exon skipping therapies in DMD," signaling a reevaluation of accelerated approvals. This cautious pivot, while protecting patients, has frustrated companies expecting streamlined paths.
Sector reactions on platforms like X (formerly Twitter) and financial forums echo devastation for patients awaiting therapies, but also calls for innovation. Analysts note that while short-term pain is evident, the gene therapy market's projected growth to $51.3 billion by 2034 could reward resilient players.
A Turning Point: Challenges for Patients, Investors, and Innovators
This wave of setbacks is devastating not just for biotechs and investors but for patients and families desperate for breakthroughs. Conditions like DMD affect mobility and lifespan, with current treatments offering limited relief. The FDA's caution is understandable—past accelerated approvals have led to post-market issues, as with Sarepta's therapies—but it risks slowing progress in a field where time is critical.
Solid Biosciences' strategic delay to gather more mature data may prove wise, allowing for stronger evidence against competitors. Looking ahead, the sector could rebound if companies adapt: emphasizing hard clinical endpoints, robust safety data, and collaborative FDA dialogues. Investors should broaden their lens—tracking competitors like Dyne and PepGen, FDA trends on surrogates, and emerging guidelines for gene-editing platforms.
Ultimately, this isn't a "dark age" but a maturation phase. With the nucleic acid and gene therapy market expanding rapidly, innovative solutions from firms like Solid Biosciences could still deliver hope. As Dr. Tidmarsh's comments remind us, the goal is therapies that truly benefit patients, not just meet minimal thresholds.
