
CGTX: Betting Against the Odds – Why Weak P-Values Won't Stop This Alzheimer's Story
When a company with FDA-aligned Phase 3 plans and a clean safety profile trades below the value of its NIH grants, something is mispriced.
The Inconvenient Truth About SHINE
Let me be blunt about something most investors gloss over: the SHINE trial's modified Intent-to-Treat (mITT) population failed. Not marginally – it failed convincingly with poor p-values that would normally send any biotech stock into oblivion.
But here's where it gets interesting.
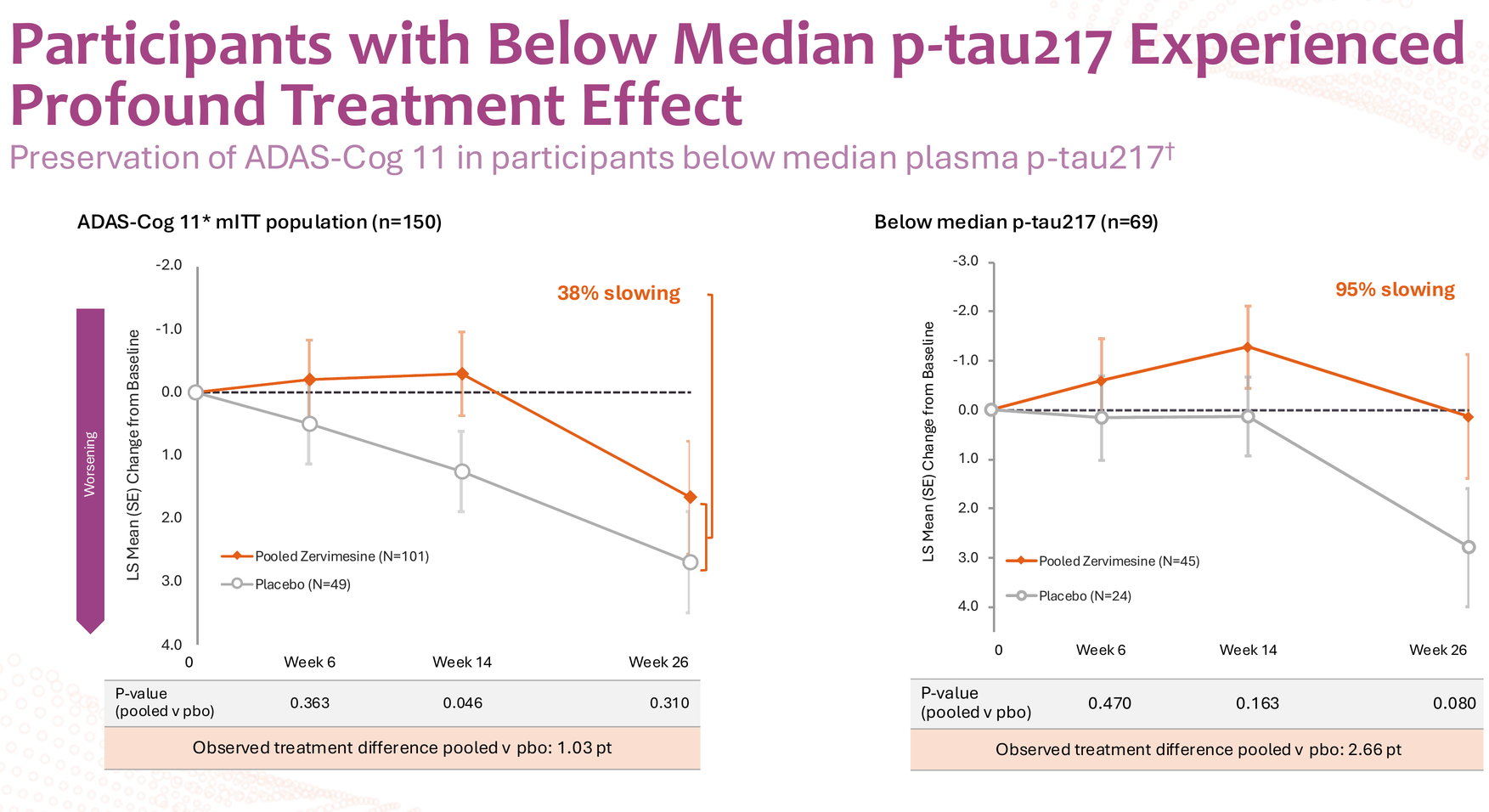
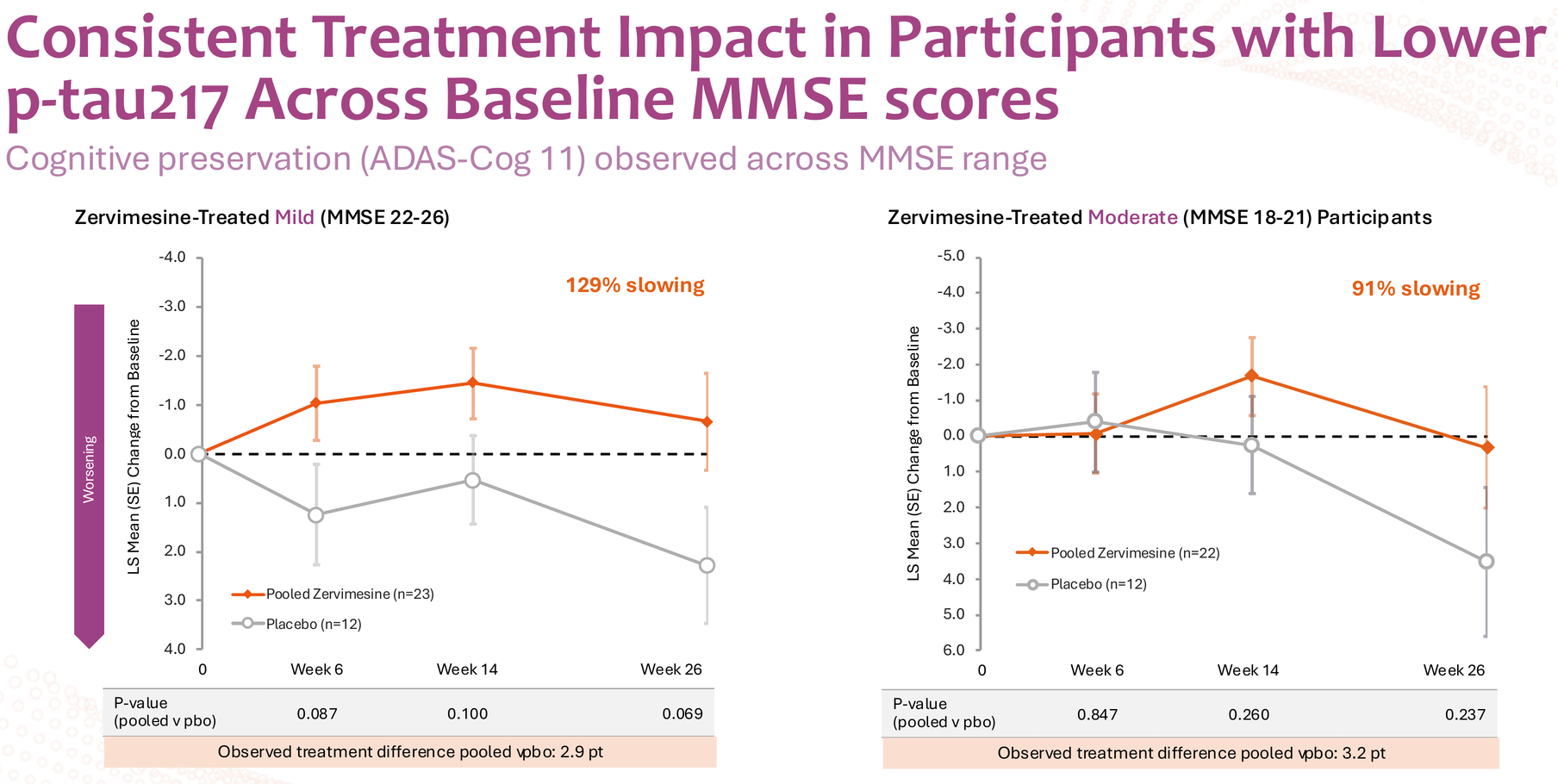
When Cognition Therapeutics sliced the data by median baseline plasma p-tau217 levels, something remarkable emerged. The below-median group showed up to 95% slowing on ADAS-Cog 11, with consistent effects across both mild (MMSE 22-26) and moderate (MMSE 18-21) disease severity – 129% and 91% slowing respectively. These aren't incremental improvements; these are disease-modification-level numbers.
The Statistical Elephant in the Room
Now for the uncomfortable part: those p-values. Ranging from 0.069 to 0.237 across the below-median subgroup analyses, these numbers violate every rule of statistical significance we learned in Biostatistics 101. In a normal world, you'd dismiss this data as noise and move on.
So why am I still betting on CGTX?
Understanding What "Below Median P-Tau217" Really Means
Here's the critical context most analysts miss: there's no naturally occurring "low p-tau217" group in Alzheimer's patients. Everyone with AD has elevated p-tau217 – that's part of the pathology. The SHINE trial simply split its 150-participant cohort at the median (1.0 pg/mL), creating a below-median group (n=69; treated n=45, placebo n=24) and an above-median group post-hoc.
This wasn't a pre-specified enrichment strategy. It was exploratory analysis that revealed a biological signal the company hadn't specifically designed the trial to detect.
And yet, the FDA paid attention.
Why the FDA Didn't Dismiss the Data
From the August 2025 press release following the End-of-Phase 2 meeting:
"In the meeting minutes, FDA concurred with our plan to enrich the Phase 3 study population with Alzheimer's patients who have lower p-tau217," stated Dr. Anthony Caggiano, Cognition's CMO. "Because p-tau217 can be measured by a simple blood test, we expect this strategy will ease the burden on patients. This enrichment strategy may increase the power of the study and reduce trial costs."
We need to read that carefully. The FDA didn't just allow an enrichment strategy – they explicitly concurred with it based on Phase 2 subgroup data that didn't meet traditional statistical thresholds.
Why?
Because when you see 2.66 points of separation on ADAS-Cog 11 at 6 months in a subgroup that represents roughly half of your AD population, that's not statistical noise – that's biological signal breaking through inadequate study design.

The Six-Month Gambit
CEO Lisa Ricciardi revealed the most important detail in that same press release:
"One of the most important outcomes from the meeting was the FDA's view that two six-month Phase 3 studies could support a new drug application for zervimesine. This plan allows us to enroll faster, more cost-effective studies, and brings us to a regulatory filing sooner."
Two parallel six-month Phase 3 trials. This is both CGTX's biggest opportunity and biggest risk.
The opportunity: SHINE already demonstrated the treatment effect at 6 months in the below-median p-tau217 group. If that signal is real, two properly powered, enriched studies should replicate it with strong statistics.
The risk: Six months is an aggressive timeline. Most AD trials run 18 months. Donanemab and lecanemab needed longer to prove their effects convincingly. If CGTX is wrong about the 6-month timeframe, both Phase 3 trials could fail simultaneously.
But here's why I think the company isn't being reckless: they have access to patient-level data we don't. They can see exactly which p-tau217 thresholds correlated with response. They can model different enrichment scenarios. And they apparently convinced the FDA that 6 months is sufficient.
The Cherry-Picking Question
Let's address the obvious criticism: isn't this just cherry-picking data to design a Phase 3 that can't fail?
Yes – and that's exactly what smart drug development should look like.
The entire concept of precision medicine is identifying the subset of patients who will benefit from a treatment. P-tau217 is simply a biomarker of disease stage and tau pathology burden. Lower p-tau217 likely indicates earlier pathology or slower disease progression – precisely the patients who might benefit most from an anti-oligomer therapy that prevents further damage rather than clearing existing plaques.
The donanemab precedent is instructive here. Eli Lilly's TRAILBLAZER-2 trial showed 36% slowing in the low-tau tertile versus 21% in the high-tau tertile. The FDA approved donanemab based partly on this tau-stratified benefit.

CGTX is taking this strategy further by prospectively enriching for likely responders. That's not cheating; that's learning from biology.
The Unanswered Questions
I'm not blind to the risks here. Several critical questions remain:
- What's the primary endpoint? SHINE showed ~2.7 points at 6 months in the below-median group. Will the Phase 3 power for 2.0 points? 1.5 points? Being too ambitious killed many AD programs. Being too conservative wastes the opportunity.
- How many patients per trial? With enrichment and strong effect sizes, these could be relatively small trials (200-250 per arm). But the company hasn't disclosed this yet.
- Will six months really be enough? This is my biggest concern. Yes, SHINE showed separation at 6 months. But will the data be convincing enough for FDA approval without longer-term data?
Despite these uncertainties, the fact that CGTX got FDA concurrence on the fundamental design gives me confidence they're not flying blind.
DLB: Promise Without Clarity
The SHIMMER trial in dementia with Lewy bodies showed intriguing signals across multiple domains – 86% slowing on NPI (neuropsychiatric inventory), 114% improvement in caregiver distress, and meaningful effects on cognition, function, and motor symptoms.

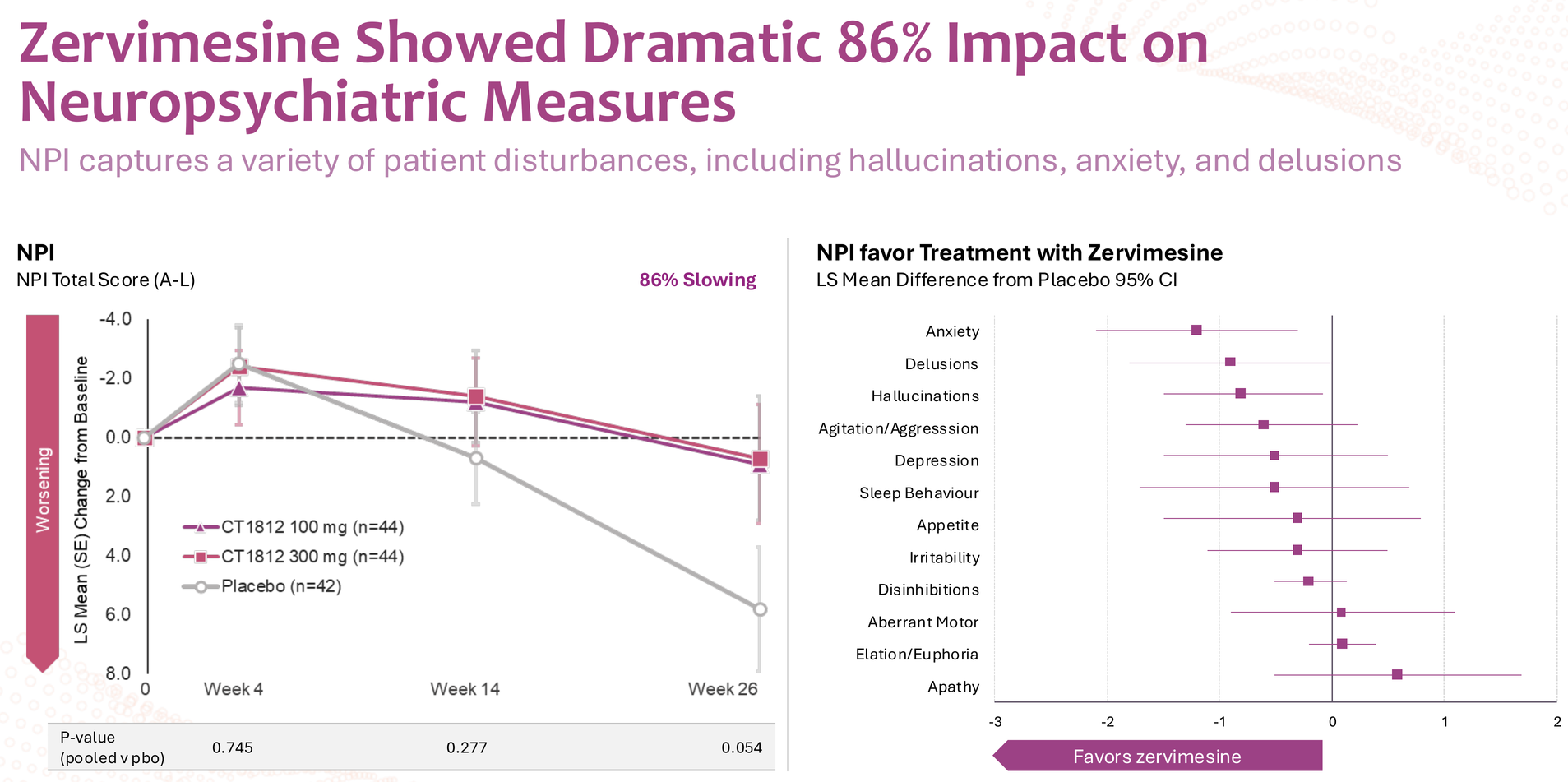
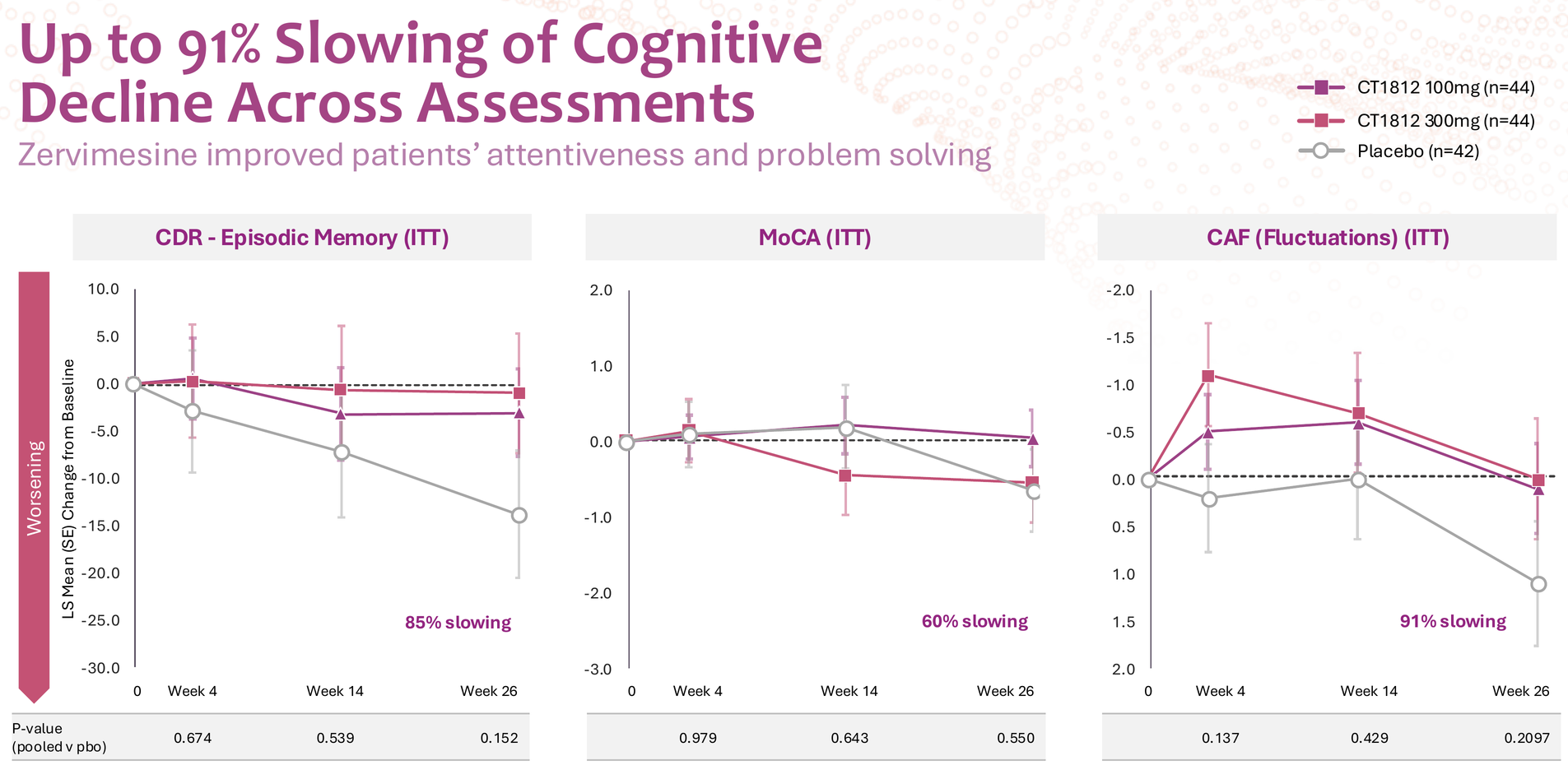
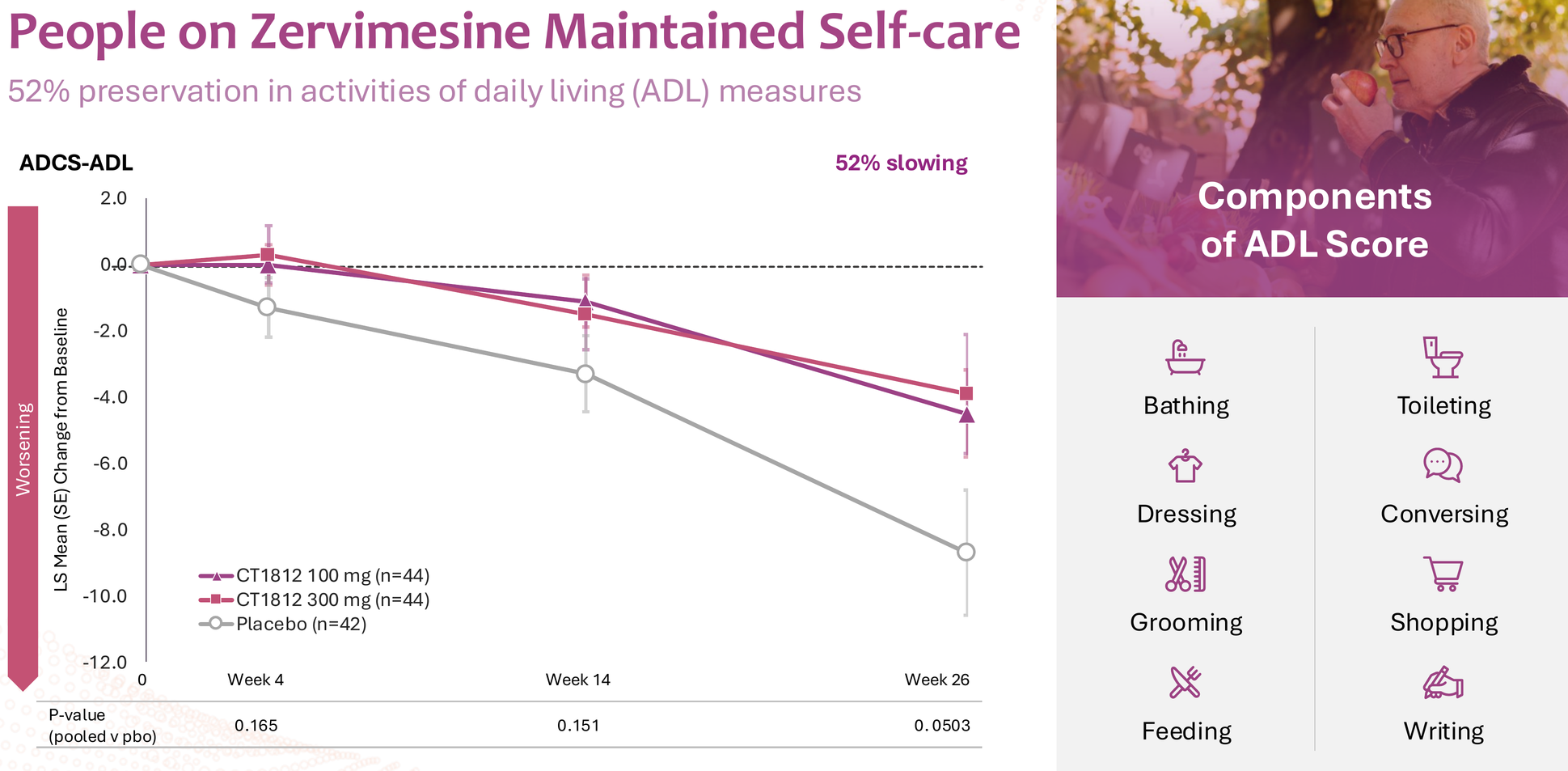
But let's be honest: this data is noisy. P-values ranged from 0.054 (NPI distress) to 0.550 (MoCA), and DLB itself is notoriously difficult to diagnose accurately pre-mortem. The clinical heterogeneity of DLB – with its fluctuating cognition, visual hallucinations, parkinsonism, and REM sleep behavior disorder – makes endpoint selection particularly challenging. For these reasons, BTD was not granted, and the FDA decided it needs to see clearer data from further studies.

CGTX will need a carefully designed Phase 2b to clarify which endpoints matter most and which patient populations respond best. The ongoing Expanded Access Program (EAP) should provide valuable real-world data, and the philanthropic donations funding it suggest genuine patient benefit beyond what clinical trial measures capture.
But scientifically, DLB represents an uphill path. Success here would be transformative – DLB is the second most common dementia with no approved disease-modifying treatments – but it's not the asset driving my investment thesis.
MAGNIFY: The Hidden Jewel
Here's what almost everyone is missing: the geographic atrophy data might be CGTX's most compelling story.
The MAGNIFY trial was terminated early due to funding constraints – a casualty of prioritizing NIH-funded AD and DLB programs over company-funded indications. But before termination, it generated remarkable signals:
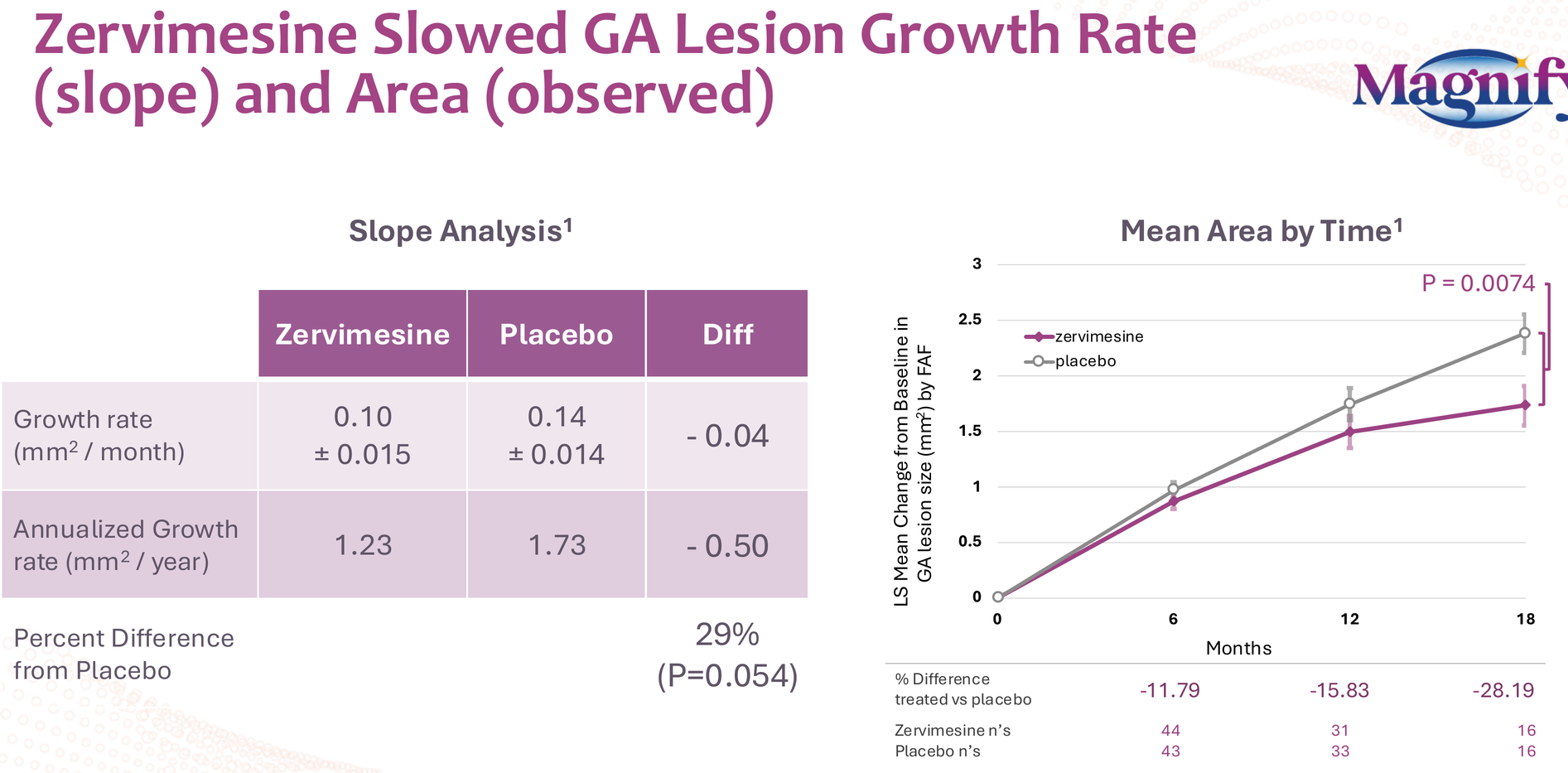
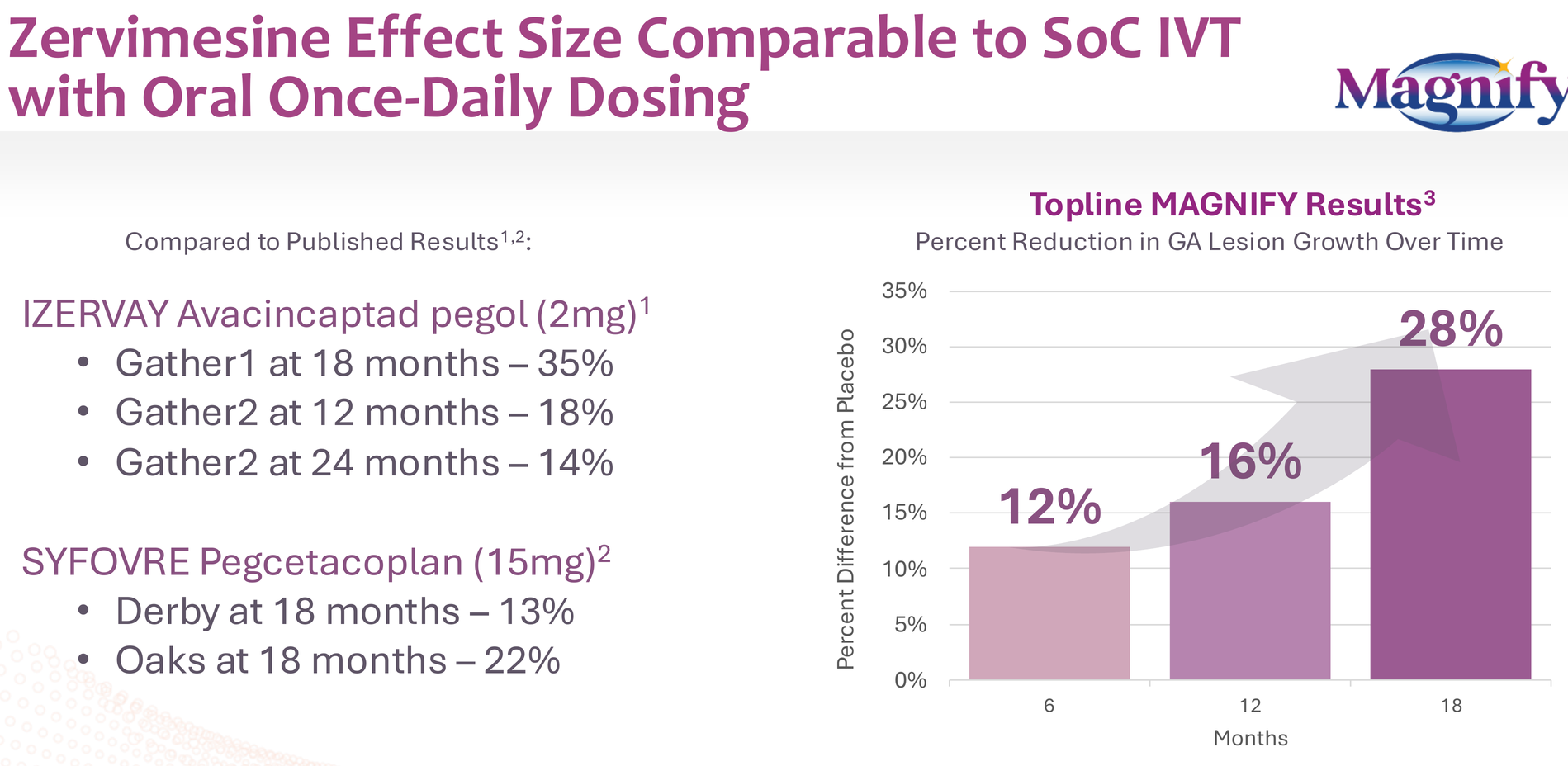
- 29% reduction in GA lesion growth rate (p=0.054)
- Effect size increasing over time: 12% at 6 months, 16% at 12 months, 28% at 18 months (p=0.0074)
- All this from an oral, once-daily pill versus the intravitreal injections required for approved therapies (Syfovre, Izervay)

Think about the patient burden difference: monthly eye injections versus a daily pill. Think about the compliance implications. Think about the market opportunity if efficacy is even comparable to approved therapies.
The trial only had 87 participants at baseline, dropping to 32 by 18 months due to early termination. With proper powering and full 24-month data, this could have been a registration-enabling trial.
This data alone should attract serious partnership interest from ophthalmology-focused pharma. The GA market is measured in billions, and an oral option would be transformative.
The Financial Reality Check
Let me be direct about the financial situation: CGTX closed a $30M equity raise in August 2025, leaving them with $11.6M in cash as of June 30, 2025 (pre-raise). They have ~$42M in remaining NIH grant funding for their trials, but that money is earmarked for specific studies and can't be freely redeployed.
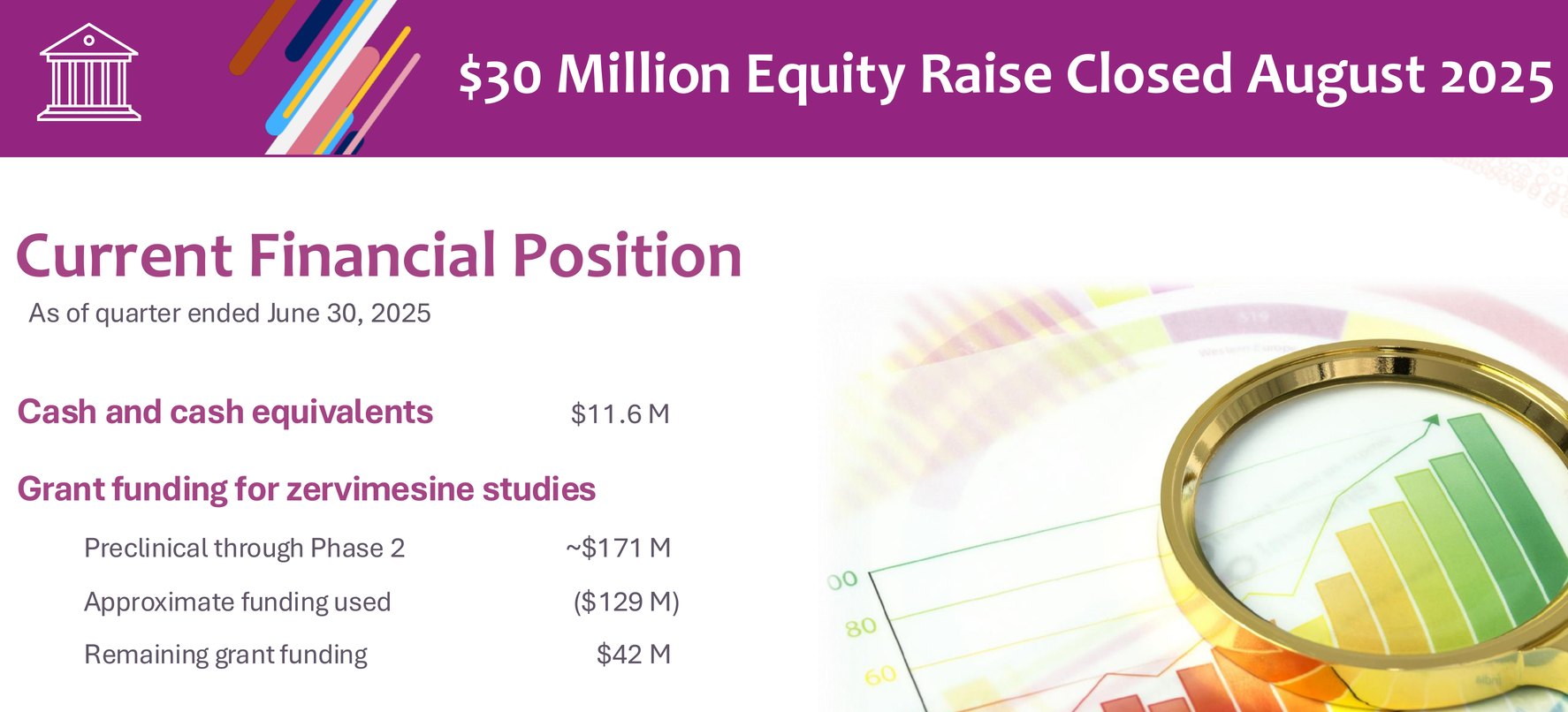
Without a partnership in the next 6-12 months, significant additional dilution is inevitable. Phase 3 AD trials will require substantial capital, even if they're "faster and more cost-effective" than traditional designs.
This is a binary bet: either the Phase 3 program attracts partnership/funding based on protocol strength, or existing shareholders face 50%+ dilution to fund development.
I'm betting on the former, but I'm not blind to the latter risk.
Why I'm Still Long
Given all the risks I've outlined – weak Phase 2 p-values, aggressive 6-month Phase 3 design, uncertain endpoints, dilution overhang – why am I maintaining and planning to add to my position?
Several reasons:
1. The FDA signal is real. Regulatory agencies don't concur with enrichment strategies and abbreviated trial designs based on noise. They saw something in the SHINE data that convinced them this was worth pursuing.
2. The biology makes sense. CT1812's mechanism – displacing oligomers from the sigma-2 receptor complex – addresses a fundamental pathogenic mechanism in AD. The overlap of amyloid-beta and alpha-synuclein pathology in both AD and DLB explains why the drug shows effects in multiple dementias.

3. The safety profile is clean. Over 450 people treated across trials with no ARIA (amyloid-related imaging abnormalities), no serious safety signals, and a side effect profile dominated by mild GI symptoms and manageable transaminase elevations in the 300mg group. The 100mg dose – what's going to Phase 3 – has been particularly clean.

4. The risk-reward is asymmetric. At an enterprise value under $120M (at time of writing), the market is pricing in near-certain failure. But if Phase 3 succeeds – and I'd put those odds at 60-70% given the totality of evidence – this becomes a multi-billion-dollar asset in a market desperate for oral AD therapies with manageable safety profiles.
5. Multiple shots on goal. Even if AD disappoints, DLB and GA provide option value. The MAGNIFY can bring a surprise gift to investors based on its available data.
My Investment Thesis
I entered CGTX at $0.65 per share. Following the EOP2 meeting with FDA, I maintained my position and plan to add once the Phase 3 protocol details are announced.
My price targets (on an enterprise value basis):
- Phase 3 initiation: $500M EV (~3.5x from current)
- Near data readout : $1B EV (~7x from current)
(will phase 3 have interim report?) - Positive Phase 3 results: $3-5B EV (partnership/acquisition territory)
These aren't based on complicated DCF models. They're based on simple comparables: what the market pays for de-risked AD assets with clean safety and oral delivery.
For context, Karuna Therapeutics was acquired by Bristol Myers Squibb for $14B based on a schizophrenia drug with promising Phase 3 data, and Alzheimer's is a far larger market.
The Partnership Question
The elephant in the room is whether CGTX can or should partner before Phase 3 data.
Arguments for early partnership:
- Eliminates dilution risk
- Provides capital for optimal trial execution
- Brings big pharma expertise in late-stage AD trials
- Validates the science
Arguments for going it alone:
- Retain maximum value for shareholders
- Prove the concept in Phase 3 before negotiating from strength
- Maintain control over trial design and execution
- Multiple assets (AD, DLB, GA) could attract better terms post-Phase 3
My read: CGTX will explore partnerships actively but won't accept lowball offers. If they can secure non-dilutive capital (debt, royalty financing) to reach Phase 3 interim data, they'll do that. If a strategic partner offers meaningful upfront + rich milestones while letting CGTX retain US rights or co-commercialization, they'll consider it.
The MAGNIFY data could be CGTX's surprise asset – the one that attracts a significant ophthalmology partnership while flying under most investors' radar. The GA market is measured in billions, and a major ophthalmology player could fund complete GA development while allowing CGTX to maintain focus and capital for their CNS programs. It's the kind of deal structure that could solve the funding equation elegantly: non-dilutive capital for AD/DLB development in exchange for GA rights, creating a win-win scenario that de-risks the entire platform.
Final Thoughts
I'm not calling CGTX a sure thing. The Phase 2 data has real statistical weaknesses. The Phase 3 design is aggressive. The balance sheet is precarious. Any number of things could go wrong.
But I've learned over years of biotech investing that the market frequently misprices binary risk. When a company with:
- A novel mechanism addressing validated pathology
- Reproducible signals across multiple diseases
- FDA alignment on a registration path
- A clean safety profile
- An oral, once-daily formulation
...is trading at an enterprise value that doesn't even cover the value of their NIH grant funding, something is disconnected from reality.
Either the science is wrong – possible, but the FDA doesn't seem to think so – or the market is underestimating probability of success.
I'm betting on the latter.
The next 18 months will tell us whether below-median p-tau217 enrichment was brilliant precision medicine or elaborate curve-fitting. Whether six months is long enough to prove disease modification. Whether CT1812's mechanism translates from Phase 2 signals to Phase 3 success.
But at current valuations, I don't need to be certain. I just need the probability of success to be materially higher than the market is pricing.
And based on everything I've analyzed, it is.
Disclosure: I own shares of CGTX with an entry cost of $0.65. I plan to add to my position upon the details of Phase 3 protocol disclosure. This is not investment advice. Do your own research. Biotech investing is inherently risky, and you can lose 100% of your investment.
This concludes my three-part analysis of Cognition Therapeutics. The journey from mechanism to market is long and uncertain, but sometimes the best opportunities hide in the complexity that others find too difficult to analyze. Time will tell if this is one of them.
Disclaimer
The information provided on this website is for informational purposes only and should not be construed as
financial, investment, legal, or professional advice. While efforts are made to ensure accuracy, no guarantee
is given regarding completeness or reliability. Visitors should conduct their own research or consult a qualified
advisor before making any decisions. External links are provided for convenience and do not imply endorsement.


