
Targeted Protein Degradation: The Drugs That Destroy Disease Instead of Blocking It — And the Biotechs Positioning to Win
The first PROTAC just got approved. Here's the science behind protein degradation, the money pouring in, the rival approaches, and the biotechs worth watching.

For forty years, almost every drug ever made worked the same way: find a protein driving disease, jam something into its active site, and hope it stays there. But roughly 80% of the proteins that cause human disease have no pocket to jam. They were written off as "undruggable." A new class of medicine ignores the pocket entirely — it tags the entire disease protein for destruction and lets the cell's own garbage disposal shred it. On 1 May 2026, the first one was approved. Here's the science, the money trail, the rival approaches, the companies worth watching — and the risks nobody puts on the slide.
The 80% Problem That Defined Modern Pharma
Think about how a normal drug works. A small molecule or an antibody finds its target protein, slots into a groove on its surface — the active site — and physically blocks it from doing its job. As long as the drug is sitting in that groove, the protein is silenced. The moment the drug washes out of the bloodstream, the protein wakes back up and resumes business.
This is occupancy-driven inhibition, and it has produced essentially every blockbuster of the modern era — the statins, the kinase inhibitors, the checkpoint antibodies. It works. But it carries three structural flaws that have quietly capped what medicine can reach.
First, the target needs a pocket. Inhibitors need a deep, well-shaped groove to bind. An estimated 80% of disease-causing proteins don't have one — transcription factors, scaffolding proteins, and large featureless surfaces like the KRAS GTPase present nothing for a drug to grab. For decades these were simply labelled "undruggable" and abandoned.
Second, blocking invites resistance. When you continuously inhibit a protein, the cell often just makes more of it. Same drug, more target, eroding effect — this is one of the most common ways cancers escape their best therapies.
Third, blocking the enzyme doesn't dismantle the scaffold. Some proteins cause disease not through their enzymatic activity but through their physical structure — acting as a docking station that holds other signalling proteins together. You can switch off the enzyme and the scaffold keeps the disease running.
For a long time, that 80% was a no-go zone. Then a chemist at Yale asked a different question: what if we didn't block the protein at all — what if we just threw it away?
The Idea: Stop Blocking, Start Shredding
Every one of your cells runs a continuous quality-control system. Proteins that are old, misfolded, or no longer needed get tagged with a small marker called ubiquitin, and that tag acts as a death sentence — the tagged protein is dragged to the proteasome, the cell's molecular wood-chipper, and destroyed. The enzymes that apply the tag are called E3 ligases, and the human body has more than 600 of them.
Targeted protein degradation (TPD) is the art of hijacking that system. Instead of blocking a disease protein, you trick the cell into tagging it for the bin. You don't occupy the target. You delete it.
The flagship version of this idea is the PROTAC — a PROteolysis TArgeting Chimera. Picture a dumbbell: one end grabs the disease protein, the other end grabs an E3 ligase, and a flexible chemical linker holds the two together. When a PROTAC clamps both ends at once, it forces the disease protein and the cellular shredding machinery into the same room. The E3 ligase tags the target with ubiquitin, the proteasome destroys it — and then comes the part that makes the whole modality so powerful:
The PROTAC walks away intact and does it again.
It isn't consumed in the reaction. A single molecule can tag one disease protein after another, catalytically. This is why degraders can work at far lower doses than inhibitors, and why the effect doesn't end the instant blood levels dip — the protein has to be physically rebuilt from scratch before the disease can resume. And because you've destroyed the entire protein rather than plugging one groove, you wipe out its enzymatic function and its scaffolding function in a single stroke. No inhibitor can do that.
That's the theory. On 1 May 2026 it stopped being theory.
The Moment It Became Real: VEPPANU
For years, sceptics had a ready dismissal of PROTACs: nice biology, but you'll never turn a two-headed, 800-plus-Dalton molecule into a pill a patient can actually swallow and absorb.
Then the FDA approved one.
VEPPANU (vepdegestrant), developed by Arvinas and Pfizer, became the first PROTAC ever approved by any regulator — cleared on 1 May 2026, more than a month ahead of its scheduled decision date, for ER-positive, HER2-negative, ESR1-mutated advanced breast cancer that has progressed after endocrine therapy. It's an oral tablet that degrades the estrogen receptor, the protein driving the cancer.
The commercial story is genuinely mixed — the pivotal VERITAC-2 trial showed a real but modest progression-free survival benefit in the mutated subgroup (5.0 months vs 2.1 for the old injectable standard), the all-comer population missed significance, and the label landed narrow. Arvinas and Pfizer ultimately handed commercialisation to Rigel Pharmaceuticals in a deal worth up to $445 million plus royalties. As a product, VEPPANU is a single asset in a crowded niche — the third drug to reach the US market specifically for ESR1-mutant breast cancer, after Menarini's Orserdu and Lilly's Inluriyo.
But that misses the point entirely. VEPPANU is infrastructure, not just a drug. Its approval proves four things the entire field needed proven: that an oral PROTAC can be formulated as a real tablet; that these ungainly molecules distribute through the human body; that grabbing both a target and an E3 ligase works in vivo; and that the FDA finds the safety profile acceptable. Every degrader behind it — in cancer, immunology, and the brain — just inherited that de-risking. The first one through the wall always takes the worst of it.
Not All Degraders Are PROTACs: The Approaches
This is where it pays to understand the engineering, because "protein degradation" is not one technology — it's a family, and the design choices separate the winners from the write-offs.
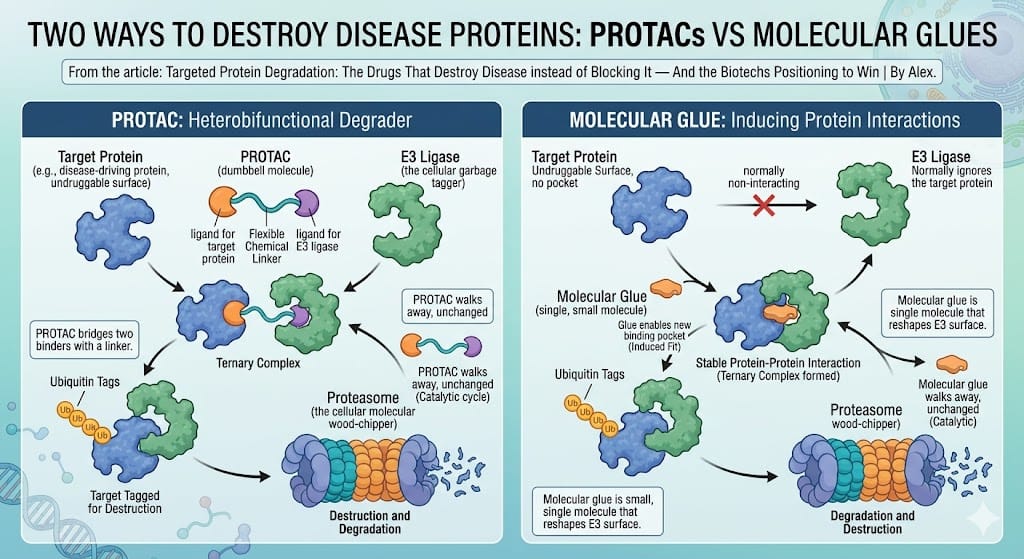
Two ways to destroy a disease protein. The PROTAC bridges target and E3 ligase with a linker; the molecular glue is a single small molecule that reshapes the E3 surface to recruit an otherwise-ignored target. Both are catalytic — the degrader is released intact and goes again.
PROTACs (heterobifunctional degraders). The dumbbell described above — two binders joined by a linker. Strengths: you can rationally design one for almost any target that has even a shallow binding site. Weakness: they're big, which makes oral absorption and tissue penetration hard. This is Arvinas's home turf.
Molecular glues. A much smaller, single molecule that doesn't bridge two binders with a linker — instead it subtly reshapes the surface of an E3 ligase so that it suddenly recognises a disease protein it would normally ignore. Think of it less as a dumbbell and more as a dab of glue at the interface. The upside is enormous: glues are small, drug-like, orally friendly, and can reach targets with no pocket at all. The catch is they've historically been hard to design on purpose — the famous ones (thalidomide derivatives) were discovered by accident. The companies that can engineer glues rationally — Monte Rosa, Kymera, Nurix — are chasing one of the most valuable capabilities in the sector.
The E3 ligase you choose. Almost every clinical degrader recruits one of just two of the body's 600-plus E3 ligases: cereblon (CRBN) or VHL. That's a crowding problem and a resistance problem (more on that below). The frontier is expanding the toolkit — discovering and drugging novel E3 ligases, some of which are expressed only in certain tissues, opening the door to degraders that act in the tumour but spare healthy organs. Nurix has built much of its identity on this E3 ligase discovery engine.
Beyond the proteasome. A wave of next-generation ideas extends the concept to proteins the proteasome can't reach: LYTACs degrade proteins outside the cell or on its surface by routing them to the lysosome; AUTACs harness autophagy; degrader-antibody conjugates bolt a degrader onto an antibody to deliver it selectively to a tumour. Most of this is still preclinical — but it's where the platform optionality lives.
The investment translation: a company with a rationally-engineered glue platform and proprietary novel E3 ligases has a far wider, more defensible opportunity than one shipping a single CRBN-based PROTAC against a single target.
Follow the Money: Big Pharma Has Already Decided
You don't have to take the science on faith. Watch where the cheques are going.
Targeted protein degradation has become one of the most heavily partnered modalities in the industry — and crucially, the deals are weighted toward upfront commitment and molecular glues, the harder, more valuable end of the field.
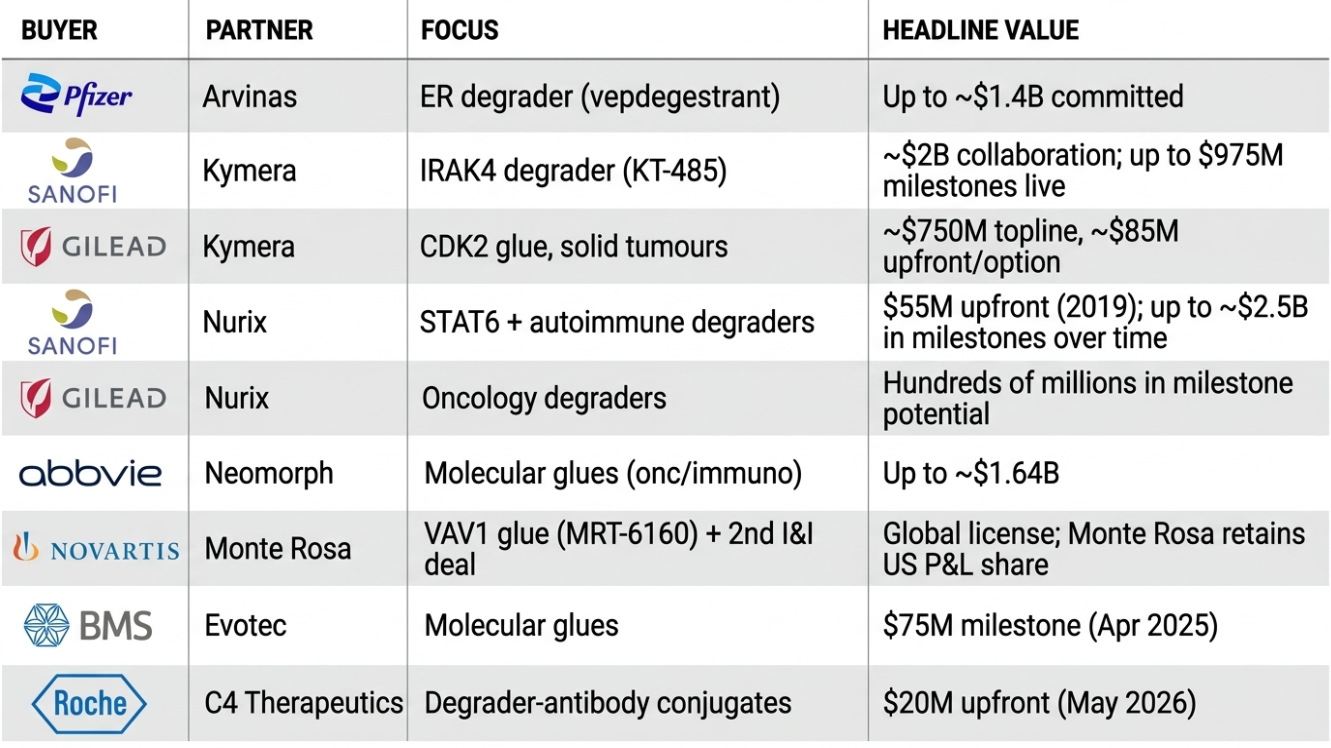
Source: company reports and announcements; data as of May 2026.
Three patterns matter. The upfronts are conviction, not options — Pfizer put roughly $1.4B behind a single degrader before approval. The smart money is buying glue platforms, not single molecules — Sanofi, Gilead, Novartis, AbbVie and BMS are all paying for the capability to make degraders, which tells you where they think the durable value sits. And the deals fund the biotechs' independence — Kymera, Nurix and Monte Rosa are each running internal pipelines on partner cash, which materially de-risks their balance sheets.
The Risk Register: What the Slide Decks Skip
Every compelling modality has an asterisk. Here's the honest accounting.
The Hook Effect. This is the one that makes degrader trials genuinely tricky. A normal inhibitor follows a clean dose-response curve — more drug, more effect, until saturation. A PROTAC needs to form a three-body complex (target + PROTAC + E3 ligase). Push the dose too high and you get the opposite of what you'd expect: PROTAC molecules saturate the target and the ligase separately, so they never get bridged together, and degradation collapses. The dose-response curve is bell-shaped, not a staircase. You cannot simply escalate to the maximum tolerated dose the way oncologists are trained to — the therapeutic window is a window, with a ceiling as well as a floor. This makes dose-finding a precision exercise, and a botched PK model shows up as erratic clinical responses, not just tolerability noise.
Oral bioavailability. Every oral degrader is a formulation feat. These molecules routinely break the medicinal chemist's rules of thumb — too big, too greasy, too poorly absorbed on paper. Patient-to-patient variation in gut absorption is a real and persistent risk.
E3 ligase resistance. Degraders elegantly beat target-level resistance — but cancer is adaptive. Cells can simply switch off the E3 ligase you're recruiting (silencing CRBN or VHL) or mutate its binding pocket, and your degrader loses its anchor. Over-reliance on the same two ligases across the whole field makes this a shared vulnerability — and is exactly why novel-E3 platforms matter.
The solid-tumour monotherapy illusion. This has bitten the field repeatedly. A degrader shows beautiful "proof of mechanism" — biopsies confirm the target protein is being destroyed — but the tumours barely shrink, because late-line solid cancers have too many escape routes for any single-agent to control. C4 Therapeutics' BRAF degrader CFT1946 is the cautionary tale: clean degradation, clean safety, and only two responses in 27 patients, leading C4 to shelve it internally and pivot. Proof of mechanism is not proof of cure.
Crowding. STAT6, IRAK4, CDK2, BTK — the most attractive targets now have multiple degrader programmes racing for them. Being first and best-tolerated will matter more than being merely present.
Biotechs Worth Following in the Degrader Space
Tier 1: Clinical-Stage Pure Plays
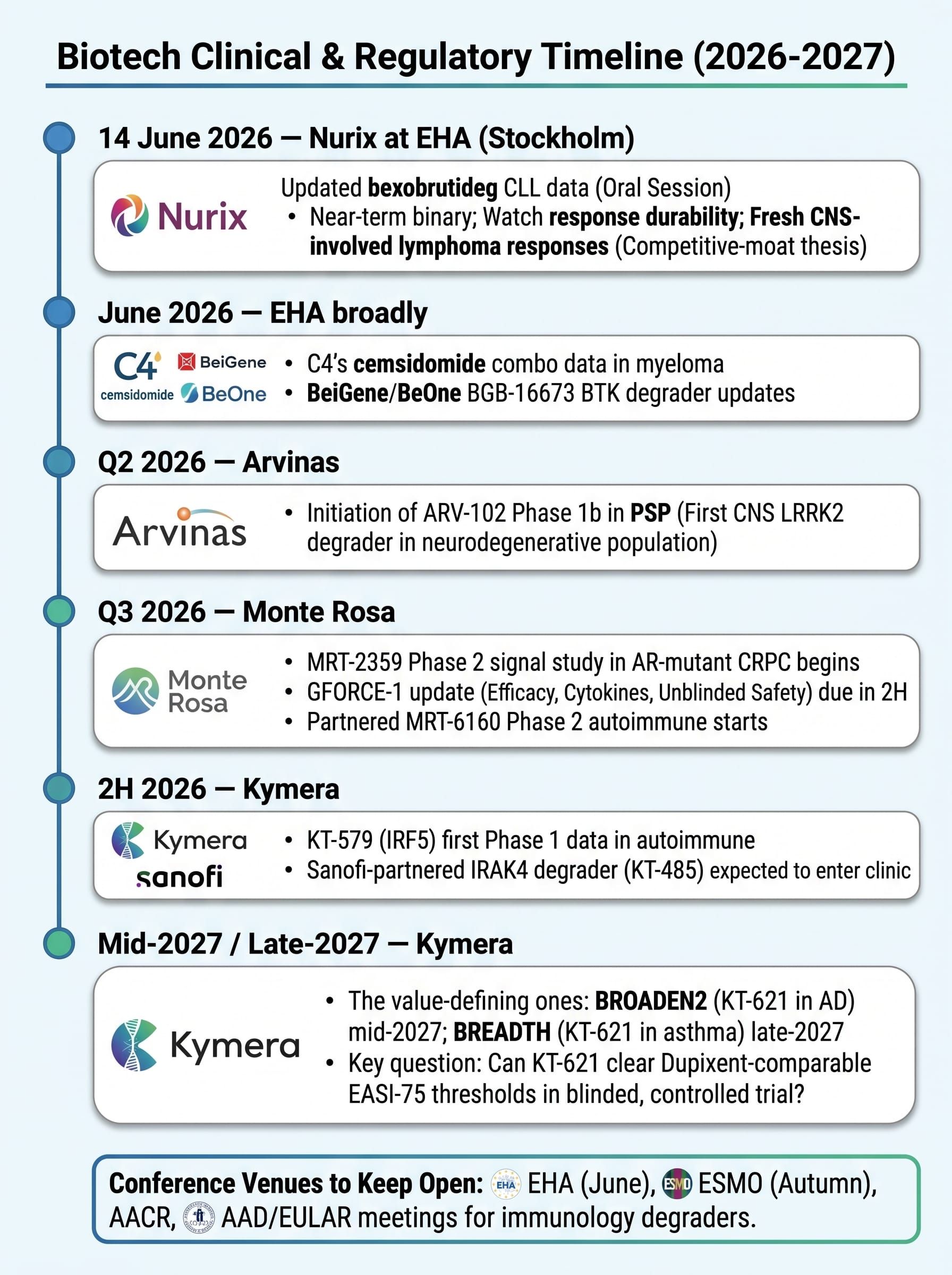
Arvinas (NASDAQ: ARVN) — The one that broke the wall. Owns the first-ever approved PROTAC (VEPPANU) and, more importantly, the platform credibility that comes with it. The real story now is ARV-102, an oral brain-penetrant PROTAC degrading LRRK2 — a target littered with the corpses of failed Parkinson's programmes that all died of fatal lung toxicity. Early Phase 1 data showed ARV-102 crossed the blood-brain barrier, degraded LRRK2 centrally, and — critically — produced no pulmonary toxicity. A Phase 1b in progressive supranuclear palsy (PSP), a uniformly fatal disease with zero disease-modifying options, is targeted to start in Q2 2026. ~$615M cash, runway into 2H 2028. The market prices ARVN as a struggling single-product launch; the bull case is that it's a de-risked CNS platform priced as if ARV-102 doesn't exist.
Kymera Therapeutics (NASDAQ: KYMR) — The cleanest risk-reward in the group and the closest thing to a fortress balance sheet. Lead asset KT-621 is a once-daily oral degrader of STAT6 — the master switch of Type 2 inflammation and the target behind Dupixent, a franchise that did over $14B in 2025. STAT6 is a transcription factor with no druggable pocket; only degradation can touch it. Early data showed deep STAT6 knockdown (>90% in early studies) and atopic-dermatitis efficacy that compares favourably to Dupixent's early benchmarks — as a pill against an injection. The value-defining readout is the BROADEN2 Phase 2b in AD, mid-2027, with an asthma readout (BREADTH) late-2027. Behind KT-621 sits an IRF5 degrader (KT-579, lupus/IBD, Phase 1 data 2H 2026) and the partnered IRAK4 degrader with Sanofi (KT-485). ~$1.55B cash, runway toward 2029 — meaning management reports its pivotal data from a position of strength, not desperation. The honest caveat: the degrader-beats-biologic thesis is still unproven in a controlled trial, and the data that settles it — whether KT-621 clears Dupixent-comparable EASI-75 thresholds in BROADEN2 — is more than a year away.
Nurix Therapeutics (NASDAQ: NRIX) — The E3-ligase house with the most concrete near-term catalyst on the board. Lead asset bexobrutideg (NX-5948) is an oral, brain-penetrant BTK degrader that destroys the entire BTK protein — beating both the kinase-domain resistance mutations and the scaffolding function that defeats conventional BTK inhibitors. Its defining edge is CNS penetrance: it has produced responses in CNS-involved lymphoma, a population systemic-only competitors structurally cannot reach. Updated CLL data is presented in an oral session at EHA in Stockholm on 14 June 2026 — the single most important near-term binary event in the degrader space. Nurix is also running a pivotal Phase 2 (DAYBreak CLL-201) and is funded by deep Gilead and Sanofi partnerships. ~$540M cash, runway toward late 2028. The risk to respect: BeOne's BGB-16673 is the more advanced rival BTK degrader, so much of Nurix's edge rests on its differentiated CNS penetrance holding up as the datasets grow.
Monte Rosa Therapeutics (NASDAQ: GLUE) — The pure-play molecular glue bet, and a different shape of risk to the PROTAC names. Its QuEEN engine is built to design glues rationally rather than stumble on them. The eye-catcher is MRT-8102, an oral NEK7 glue hitting the NLRP3 inflammasome for cardiovascular inflammation — early Phase 1 data showed an 85% reduction in hsCRP at four weeks, with most subjects driven below the 2 mg/L risk threshold. Its VAV1 glue (MRT-6160) is licensed to Novartis with Monte Rosa retaining a US profit-share and multiple Phase 2 autoimmune starts due; an oncology glue (MRT-2359) heads into a Phase 2 CRPC signal study around Q3 2026. The thesis: glues are the more drug-like, more partnerable end of TPD, and Monte Rosa is the listed way to own that.
C4 Therapeutics (NASDAQ: CCCC) — The repositioning story. After shelving its BRAF degrader, C4's lead is now cemsidomide, an IKZF1/3 molecular glue in multiple myeloma (MOMENTUM Phase 2; a Phase 1b combination with Pfizer's elranatamab; data at EHA 2026). A new Roche degrader-antibody-conjugate deal ($20M upfront, May 2026) adds non-dilutive funding. ~$268M cash, runway to end of 2028. The honest read: adequately funded through its readouts, but cemsidomide must now win in a crowded myeloma field rather than carry the original platform narrative.
Tier 2: The Large-Cap Validators
Pfizer (NYSE: PFE) — Co-owner of the first approved PROTAC; the deepest-pocketed believer, having committed ~$1.4B to vepdegestrant.
Astellas (Tokyo: 4503) — Advancing setidegrasib, a degrader of KRAS G12D — the mutation that resisted four decades of drug discovery and drives ~40% of pancreatic cancer. Phase 1 data (NSCLC ~33% ORR; PDAC activity with clean ctDNA biomarker correlation) earned a move into first-line Phase 3 PDAC. If KRAS G12D degradation works, it's one of the largest prizes in oncology.
BeiGene / BeOne (NASDAQ: ONC) — Holds the most advanced rival BTK degrader, BGB-16673, with deep responses and the commercial muscle of a global oncology franchise behind it. The incumbent threat to Nurix's CLL story.
Novartis, Sanofi, Gilead, AbbVie, BMS, Roche — Not degrader pure-plays, but each has paid up to own a piece of the platform (see the money trail). Watch them as the eventual acquirers: a clean Phase 2 from any Tier 1 name above is exactly the kind of asset this group has shown it will pay billions for.
Tier 3: Private / Pre-IPO (Not Yet Tradeable)
A watchlist for when they list or get bought. Neomorph (glues; AbbVie, BMS, Novo partners), Plexium, Seed Therapeutics, Avilar Therapeutics (LYTACs — extracellular degradation), Amphista (linker-free "targeted glues"), and Captor (structure-led precision degraders). Several are obvious M&A or IPO candidates in 2026–2027.
Catalyst Calendar: Mark These Dates
The degrader space doesn't have a single make-or-break date — instead, a steady drumbeat of real readouts, with one genuinely imminent oral presentation.
The Bottom Line
Targeted protein degradation just crossed the line from elegant theory to approved medicine, and the market hasn't fully repriced what that means for everything behind the first approval. The modality solves a problem — the undruggable 80% of the proteome — that occupancy-based drugs structurally cannot, and Big Pharma has already voted with billions of dollars, concentrated tellingly on the harder molecular-glue end of the field.
For platform exposure with a fortress balance sheet, Kymera (KYMR) offers the cleanest risk-reward — best-in-class early data, an undruggable target with no oral rivals, and runway toward 2029 that lets it report pivotal data on its own terms. For the most concrete near-term catalyst, Nurix (NRIX) has a genuine binary on 14 June. For an underappreciated CNS platform priced as a struggling launch, Arvinas (ARVN) is the contrarian setup — the value is in ARV-102, not VEPPANU. For pure molecular-glue exposure, Monte Rosa (GLUE) is the listed way to own the most partnerable corner of the field. And among the large caps, Astellas's KRAS G12D degrader is the highest-prize lottery ticket if it reads out.
The risks are real and specific: the hook effect makes dosing a precision game, oral absorption is never guaranteed, the same two E3 ligases create a shared resistance vulnerability, and solid-tumour monotherapy data has flattered to deceive before. This is binary-outcome biotech. Size positions accordingly, diversify across targets and stages, and keep your calendar clear for mid-June — because when the EHA data drops, you'll want to be paying attention.
Disclaimer: This article is for informational and educational purposes only. It does not constitute financial or investment advice. BioStockInfo receives no compensation from any company discussed. Always conduct your own due diligence before making investment decisions.



